
Zantac Litigation Overview
Article Provided by Brenden Leydon

Attorney at Wocl Leydon LLC
The litigation over the harmful effects of Zantac (generic name is Ranitidine) is just getting underway, and it looks like it will lead to a significant number of cases.
What is Zantac/Ranitidine?
Ranitidine is an antacid which belongs to a class of drugs known as histamine-2 receptor agonists (H2 blockers). This medication was also available without a prescription. It was used as a common treatment used for ulcers, heartburn, indigestion, and gastroesophageal reflux disease (GERD). It works by effectively blocking histamine receptors in the stomach lining to decrease stomach acids. These drugs work by preventing the production of stomach acid via an enzyme called the proton pump. Zantac starts relieving symptoms of heartburn and related conditions in about 30 minutes. The drug’s acid prevention benefits can last for up to 12 hours per dosage.
Ranitidine was first invented in 1977 by GlaxoSmithKline (GSK). In 1983, Zantac was approved by the FDA and became widely available in the U.S. By 1988, it was considered the world’s best-selling drug, with over $1 billion in annual sales. In 1997, GSK’s U.S. patent for Zantac expired. Competitors began manufacturing generic ranitidine drugs alongside the brand name, Zantac. In 2004, Pfizer launched the first non-prescription version of Zantac, which was sold over-the-counter (OTC). The Zantac brand was purchased from Pfizer first by Johnson & Johnson, then by Boehringer Ingelheim Pharmaceuticals, and then, most recently, by Sanofi SA. Generic over-the-counter versions were also released to the U.S. market. In 2018, Zantac was prescribed to over 25 million patients.
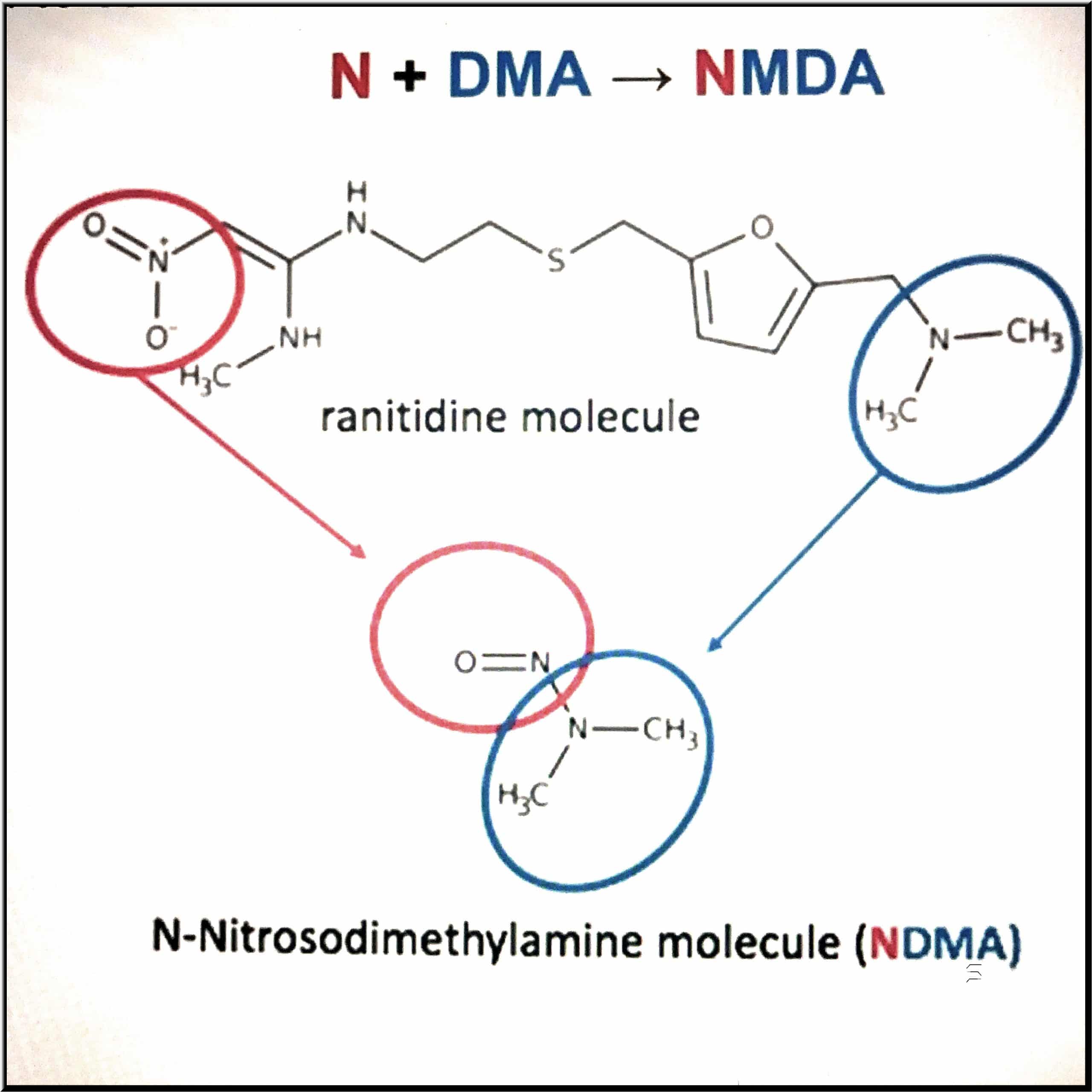
The Zantac NDMA defect
N-nitrosodimethylamine (NDMA) is a highly dangerous and potent carcinogen. It is classified as an extremely hazardous substance in the United States as defined in Section 302 of the U.S. Emergency Planning and Community Right-to-Know Act (42 U.S.C. 11002) and is subject to strict reporting requirements by facilities that produce, store, or use it in significant quantities. NDMA actually has an industrial use, which is specifically to induce tumors in animals for purposes of laboratory testing
As a known human carcinogen, the NDMA levels observed in ranitidine products presented clear cancer risks. The FDA also determined that the inherent instability of the ranitidine molecule supports NDMA formation both within the medication and within the human body upon digestion. The dangers of ranitidine drugs were publicly uncovered by Valisure, a Connecticut-based online pharmacy, in September 2019. The company informed the FDA that it found shocking levels of NDMA in the drug. The acceptable daily limit of NDMA is 96 nanograms, but Valisure found over 3,000,000 nanograms in every lot of ranitidine that it tested.

In response to this, the FDA issued a warning that some ranitidine medications may contain NDMA. The FDA also recommended that manufacturers test their products. After this announcement, certain ranitidine manufacturers and retailers suspended sales of Zantac or generic ranitidine. Then, on April 1, 2020, the FDA requested that all manufacturers withdraw prescription and over-the-counter medications containing ranitidine from the market immediately. The reason given was that the FDA had since done additional testing which showed that the amount of NDMA present in Zantac increased over time, especially when the drug was stored at high temperatures. This could result in unacceptable daily intake levels of NDMA. The FDA recommended that consumers stop taking ranitidine medications or to speak with their health care providers about safe alternatives.
Additional defect because Ranitidine is a nitrosatable drug
The FDA has mostly focused on the fact that they recently discovered (though defendants knew or should have known for at least two decades) that Zantac/Ranitidine begin converting to NDMA not only at high levels of heat, but at room temperature was well. Many drugs require refrigeration to prevent deterioration or conversion to harmful substances. These drugs must be maintained at “refrigeration” temperatures from the time they “roll off” the manufacturing line, during warehousing, shipment, at retail locations and ultimately in the consumers home (or other location). Insulins, antibiotic liquids, injections, eye drops, and some creams require constant refrigeration.
In order to prevent Zantac/Ranitidine from producing NDMA, the drug would need to have been manufactured at “refrigeration” temperatures and maintained at these temperatures by every entity in the supply chain that took possession of the product prior to purchase by the consumer. The manufacturer as well as other members of the supply chain, including retailers, were under a duty to instruct the consumer to continue to maintain the product at “refrigeration” temperatures” post purchase.
Although the temperature related production of NDMA is a significant issue, there is another equally or possibly more significant issue relevant to the production of NDMA and Zantac/Ranitidine. Nitrosatable drugs, such as secondary or tertiary amines and amides, form N-nitroso compounds in the presence of nitrites and acids. Zantac/Ranitidine are nitrosatable drugs. The N-nitroso compound formed when Zantac/Ranitidine is ingested and interacts with stomach acids and other gastric juices is NDMA. The defendants knew or should have known that Zantac/Ranitidine was a Nitrosatable drug and that the N-nitroso compound produced post consumption is NDMA.
Making a bad situation worse, when Nitrosatable drugs are exposed to stomach acids in an individual with abnormally acidic (lower Ph) stomach acids, an exponentially larger quantity of NDMA is produced as compared to individual with normal levels of stomach acidity. This is rather shocking when you consider the purpose of the drug itself is to treat high stomach acidity, thus by design maximizing this dangerous attribute. As Zantac/Ranitidine products were specifically designed for and marketed to individuals with abnormally acidic stomach acids, the product was designed for and marketed to a patient population that was at the highest level of risk from consumption of the product. The breach of duty to adequately test this product or warn about its harmful consequences is extraordinary.
Cancers potentially caused by Zantac
As noted above, the high concentrations of NDMA users of Zantac may have been exposed to are strongly associated with a variety of cancers. Considering NDMA is literally used to induce tumors in laboratory experiments, there is biological plausibility for virtually any cancer. The research on this issue is still ongoing and under development, but among the ones that look to have a strong association are:
- Bladder Cancer
- Breast Cancer
- Esophageal Cancer
- Kidney (renal) Cancer
- Liver Cancer
- Lung Cancer
- Pancreatic Cancer
- Prostate Cancer
- Testicular Cancer
- Stomach Cancer
It is likely this list will be adjusted, including adding other cancers, as more research continues.
Litigation options
After numerous federal court actions were filed throughout the country, in Spring of 2020 the Federal Court established a Multi District Litigation (MDL) for Zantac/Ranitidine in the Southern District of Florida. Many other state court actions are also pending. The MDL has a couple of options to consider. Attorneys can direct file in the MDL without a need to obtain local counsel or be admitted pro hac vice. Additionally, there is a registration option which allows preserving the claim and tolling the statute without incurring the expense of actually filing suit. This option also allows for a cost split with the defendants for obtaining certain medical records. There is an MDL website set up that has many of the relevant orders easily accessible
https://www.flsd.uscourts.gov/zantac
Another option is filing in state court. There have been cases filed in state court where various defendants are located and thus can’t remove to federal court under the forum defendant rule. For instance, Boehringer Ingelheim, headquartered in Connecticut, owned the rights to Zantac between 2006 and 2017. One matter to consider in deciding where to file are potential evidentiary considerations. One that is likely to come into play is the subsequent remedial measures doctrine. Obviously the removal of these products from the market is powerful evidence of their defective nature. Under Connecticut law (where Boehringer Ingelheim is headquartered), this subsequent remedial measure is admissible and essentially in and of itself arguably proves Plaintiff’s strict product liability case.
Under Connecticut law, although evidence of subsequent remedial measures is generally not admissible in a negligence action on the issue of whether the defendant failed to exercise due care, it is admissible in a strict liability action on the issue of whether the product was defectively designed if the remedial repairs are related to the alleged design defect.
Wagner v. Clark Equip. Co., 243 Conn. 168, 193, 700 A.2d 38, 51 (1997). Conn. Code Evid. Sec. 4-7 (“(b) Strict Product Liability of Goods. Where a theory of liability relied on by a party is strict product liability, evidence of such measures taken after an event is admissible.”). “Subsequent change evidence may be highly probative of defectiveness because it provides a safer alternative against which to compare the allegedly defective product.” Wagner, supra, 243 Conn. at 195.
However, in federal court, it has been held that Rule 407 operates to keep such evidence out even in strict liability cases. Cann v. Ford Motor Co., 658 F.2d 54, 60 (2d Cir. 1981)(“we hold that Rule 407 applies to strict products liability actions”). Courts have specifically held this to be the case even in a diversity case where the substantive law of the forum case is supposed to be applied.
However, other Circuit courts have held that Rule 407 applies when there is conflicting state law regarding the admissibility of subsequent remedial measures as it is Congress, the entity that enacted the Federal Rules of Evidence, that has the authority to decide what evidence should be excluded in diversity cases. See, e.g. Flaminio v. Honda Motor Co., Ltd., 733 F.2d 463,468-72, (7th Cir. 1984). As a result of the foregoing, the Court will apply Rule 407 in this case.
Napolitano v. Synthes, Inc., No. 09-CV-828 (TLM), 2014 WL 12868860, at *2 (D. Conn. Apr. 21, 2014). While there may be other exceptions or arguments that would allow the withdrawal of this product from the market to be admissible in a federal court action, it is much more clearly likely to be admissible in a state court if that state expressly allows the admission of this evidence.
In conclusion, it looks like this litigation will continue to grow over the next few years, with a very strong liability case to be made, which will likely only grow stronger as more research comes out.


Comments